
Vergleich der PMS zwischen China und der EU
21. April 2023In China ist die Überwachung nach dem Inverkehrbringen (Post-Market Surveillance, PMS) ein wichtiger Bestandteil der Regulierung von Medizinprodukten, um die Sicherheit und Wirksamkeit dieser Produkte zu gewährleisten. Sie wird von der NMPA (National Medical Products Administration) beaufsichtigt. Dieser Artikel gibt einen Überblick über die wichtigsten Punkte der PMS in China und vergleicht die PMS zwischen China und der EU.
Behalten Sie diese Definitionen im Hinterkopf
In China ist ein unerwünschtes Ereignis bei Medizinprodukten jedes nachteilige Ereignis, das bei der Verwendung von vermarkteten Medizinprodukten unter normalen Bedingungen auftritt und dem menschlichen Körper Schaden zufügt oder zufügen könnte.
Gemäß der MDR bedeutet „unerwünschtes Ereignis" ein nachteiliges medizinisches Ereignis, eine nicht vorgesehene Erkrankung oder Verletzung oder nachteilige klinische Symptome, einschließlich anormaler Laborbefunde, bei Prüfungsteilnehmern, Anwendern oder anderen Personen im Rahmen einer klinischen Prüfung, auch wenn diese nicht mit dem Prüfprodukt zusammenhängen;
„Vorkommnis” bezeichnet eine Fehlfunktion oder Verschlechterung der Eigenschaften oder Leistung eines bereits auf dem Markt bereitgestellten Produkts, einschließlich Anwendungsfehlern aufgrund ergonomischer Merkmale, sowie eine Unzulänglichkeit der vom Hersteller bereitgestellten Informationen oder eine unerwünschte Nebenwirkung;
Die chinesische Provisions for Medical Device Adverse Event Monitoring and Re-evaluation definiert den schwerwiegenden Schaden wie folgt:
“Serious damage is defined as the damage that one of the following situations is met:
- Life threatening;
- Causing permanent harms to body function or injuries to body structure;
- Medical measures are required to avoid the above-mentioned permanent harms and injuries.“
Die MDR definiert „schwerwiegendes Vorkommnis“ als ein Vorkommnis, das direkt oder indirekt eine der nachstehenden Folgen hatte, hätte haben können oder haben könnte:
- den Tod eines Patienten, Anwenders oder einer anderen Person,
- die vorübergehende oder dauerhafte schwerwiegende Verschlechterung des Gesundheitszustands eines Patienten, Anwenders oder anderer Personen,
- eine schwerwiegende Gefahr für die öffentliche Gesundheit;
Ein gruppenbezogenes unerwünschtes Ereignis (Group adverse event, aus Provisions for Medical Device Adverse Event Monitoring and Re-evaluation) im Zusammenhang mit einem Medizinprodukt bezieht sich auf Ereignisse, die während der Anwendung desselben Medizinprodukts auftreten und die Gesundheit oder Lebenssicherheit einer bestimmten Bevölkerungsgruppe innerhalb eines relativ konzentrierten Zeitraums und Gebiets beeinträchtigen oder gefährden.
Zulassungsinhaber für Medizinprodukte (Medical device marketing authorization holders, MAHs, aus Provisions for Medical Device Adverse Event Monitoring and Re-evaluation) beziehen sich auf die Inhaber der Registrierungsbescheinigung für Medizinprodukte und der Anmeldebescheinigung, d. h. die MAHs für Medizinprodukte und die anmeldenden Personen.

Verantwortlichkeiten der Zulassungsinhaber
Die Zulassungsinhaber sind verantwortlich für die kontinuierliche Erforschung bereits in Verkehr gebrachter Medizinprodukte, die Risikobewertung, die Überwachung unerwünschter Ereignisse im Zusammenhang mit Medizinprodukten, die Umsetzung wirksamer Kontrollmaßnahmen auf der Grundlage der Analyse- und Bewertungsergebnisse sowie die Erfüllung der folgenden Hauptpflichten:
- Einrichtung eines Qualitätsmanagementsystems für Medizinprodukte, das ein funktionierendes Verfahren zur Überwachung unerwünschter Ereignisse und zur Neubewertung von Medizinprodukten umfasst;
- Ausstattung mit der geeigneten Organisation und dem geeigneten Personal für die Durchführung der PMS-Arbeit;
- Proaktive Erfassung und wahrheitsgemäße und unverzügliche Meldung von unerwünschten Ereignissen bei Medizinprodukten an die Überwachungsbehörden innerhalb des in den Vorschriften festgelegten Zeitrahmens;
- Unverzügliche Untersuchung, Analyse und Bewertung von unerwünschten Ereignissen bei Medizinprodukten sowie unverzügliche Ergreifung von Risikokontrollmaßnahmen und Veröffentlichung von Risikoinformationen;
- Durchführung einer kontinuierlichen Studie über die Sicherheit von Medizinprodukten, sobald diese auf dem Markt sind, und Erstellung regelmäßiger Risikobewertungsberichte nach Bedarf;
- Aktive Durchführung von Neubewertungen von Medizinprodukten;
- Zusammenarbeit mit der NMPA bei der Untersuchung von unerwünschten Ereignissen bei Medizinprodukten
MAHs in Übersee sollten auch einen Mechanismus zum Informationsaustausch mit ihren benannten Bevollmöchtigten einrichten, um Informationen über die Überwachung und Neubewertung von unerwünschten Ereignissen bei Medizinprodukten auszutauschen.
PMS-Management-System
Der Zulassungsinhaber sollte ein PMS-Managementsystem einrichten, das folgende Aspekte umfasst:
- Führende Gruppe. Die Zulassungsinhaber sollten eine Führungsgruppe mit dem Verantwortlichen des Unternehmens als Teamleiter einrichten, die sich aus Mitarbeitern der Abteilungen für Forschung und Entwicklung, Produktion, Vertrieb, Kundendienst und Überwachung von unerwünschten Ereignissen zusammensetzt und die Überwachung von unerwünschten Ereignissen bei Medizinprodukten umfassend leitet, organisiert und verwaltet.
- PMS-Abteilungen und -Personal. Das PMS-Personal sollte eine professionelle Ausbildung in der Überwachung von unerwünschten Ereignissen bei Medizinprodukten absolviert haben, mit den einschlägigen Vorschriften zur Überwachung von unerwünschten Ereignissen bei Medizinprodukten vertraut sein, über professionelle Kenntnisse von Medizinprodukten verfügen, mit den Merkmalen der Medizinprodukte, die sie in der Hand haben, vertraut sein und über gute Kommunikations- und Koordinationsfähigkeiten verfügen. Das PMS-Personal ist verantwortlich für die Sammlung von Meldungen über unerwünschte Ereignisse bei Medizinprodukten und die rechtzeitige Meldung an die NMPA sowie für die Organisation der Erstellung von PMS-Arbeitsverfahren, Notfallplänen und anderen Dokumenten.
- Die PMS-Schulung sollte das gesamte Unternehmenspersonal umfassen. Zu den Schulungsinhalten sollten die aktuellen einschlägigen Gesetze und Vorschriften, PMS-Arbeitsverfahren, einschlägige Kenntnisse über PMS, Analyse- und Bewertungsmethoden sowie Kenntnisse über den rationellen Einsatz von Geräten usw. gehören. Nach der Schulung sollte der Schulungseffekt anhand von Fragebögen und schriftlichen Prüfungen bewertet werden.
- Untersuchung. Unerwünschte Ereignisse, die zum Tod oder zu schweren Verletzungen führen, oder die das Potenzial haben, schwere Verletzungen oder den Tod zu verursachen, sollten untersucht werden. Der Hauptinhalt der Untersuchung umfasst den Qualitätsstatus des Produkts, die Korrelation zwischen der Verletzung und dem Produkt usw.
- Notfallmaßnahmen. Wenn ein unerwünschtes Ereignis bei einem Medizinprodukt eine Notfallreaktion erfordert, sollte der Zulassungsinhaber rechtzeitig die erforderlichen Risikokontrollmaßnahmen ergreifen, wie z. B. die Einstellung der Verwendung, die Einstellung des Verkaufs, den Rückruf usw.
- Überwachungsaufzeichnungen. Die Aufzeichnungen sollten mindestens 2 Jahre nach Ablauf des Haltbarkeitsdatums von Medizinprodukten aufbewahrt werden; bei Medizinprodukten ohne Haltbarkeitsangabe sollte die Aufbewahrungsfrist nicht weniger als 5 Jahre betragen; die Überwachungsaufzeichnungen für implantierbare Medizinprodukte sollten dauerhaft aufbewahrt werden.

Meldung von unerwünschten Ereignissen
In China sollten unerwünschte Ereignisse nach dem Grundsatz "Im Zweifelsfall melden" gemeldet werden. Wenn der Verdacht besteht, dass es sich bei einem Ereignis um ein Medizinprodukt handelt, sollte es als solches gemeldet werden. In der EU müssen die Hersteller jedes einzelne schwerwiegende Vorkommnis melden, nachdem sie den Kausalzusammenhang zwischen diesem Vorkommnis und ihrem Produkt nachgewiesen haben oder ein solcher Kausalzusammenhang vernünftigerweise möglich ist. Im Falle einer schwerwiegenden Gefahr der öffentlichen Gesundheit ist die Meldung jedoch unverzüglich und ohne Kausalzusammenhang vorzunehmen.
Das verdächtige unerwünschte Ereignis sollte über die NMPA-Online-Plattform https://www.cdr-adr.org.cn/ylqx_1/Medical_blsjbg/ gemeldet werden. Im Ausland ansässige Hersteller von importierten Medizinprodukten und Hersteller, die inländische Medizinprodukte ins Ausland exportieren, sammeln freiwillig die im Ausland aufgetretenen unerwünschten Ereignisse und melden unerwünschte Ereignisse, die zu schweren Verletzungen oder zum Tod führen oder führen könnten, innerhalb von 30 Tagen nach ihrer Feststellung oder Meldung.
In der folgenden Tabelle werden die Meldefristen in China und in der EU verglichen.
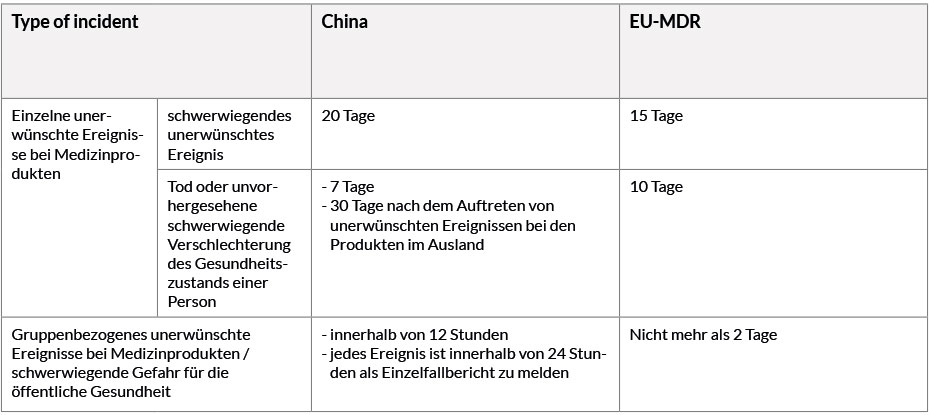
Analyse und Bewertung von unerwünschten Ereignissen
Nach der Meldung sollte der Hersteller das unerwünschte Ereignis analysieren. Zu den wichtigsten Inhalten der Bewertung gehören:
- ob das Produkt mit den verbindlichen Normen und den registrierten oder hinterlegten technischen Spezifikationen übereinstimmt;
- ob während der Verwendung des Medizinprodukts eine Fehlfunktion oder eine Verletzung aufgetreten ist;
- ob es unter normalen Anwendungsbedingungen zu Verletzungen führen würde und ob es wissenschaftliche Literatur, Untersuchungen, relevante Tests oder Überprüfungen gibt, die die Ursache der Verletzung erklären können;
- die Region und die Merkmale der von der Verletzung betroffenen Bevölkerung;
- die Schwere der Schädigung der menschlichen Gesundheit;
- die Wahrscheinlichkeit einer Verletzung;
- die kurz- und langfristigen Folgen der Schädigung;
- andere Faktoren, die eine Verletzung des menschlichen Körpers verursachen können.
Unsere Fortbildungen
Sie möchten mehr erfahren? Dann buchen Sie das passende Seminar zum Thema. Oder benötigen sie eine Inhouse-Schulung? Dann kontaktieren Sie uns gerne unter academy@qtec-group.com



Risikokontrolle
Stellt der Zulassungsinhaber fest, dass Medizinprodukte mit unangemessenen Risiken behaftet sind, die die menschliche Gesundheit und die Sicherheit gefährden können, sollte er je nach Risikosituation geeignete Kontrollmaßnahmen ergreifen, z. B. die Einstellung der Produktion und des Verkaufs des Produkts und die Benachrichtigung des betreffenden Anwenders über die Aussetzung der Verwendung, die Durchführung eines Produktrückrufs, die Herausgabe von Risikowarnungen, die Durchführung einer Selbstprüfung und die Verbesserung des Qualitätssicherungssystems der Produktion, die Überarbeitung der Kennzeichnung und der Gebrauchsanweisung, die Verbesserung des Verfahrens, des Designs, der technischen Produktanforderungen usw.
Wenn Medizinprodukte aufgrund von unerwünschten Ereignissen im Ausland Kontrollmaßnahmen unterliegen, sollte der Registrant die unerwünschten Ereignisse, Kontrollmaßnahmen und die anschließende Entsorgung in China innerhalb von 24 Stunden an die NMPA melden.
Wenn sich die Erkenntnisse über die Sicherheit und Wirksamkeit des Medizinprodukts ändern, die Überwachungs- und Bewertungsergebnisse unerwünschter Ereignisse zeigen, dass das Medizinprodukt möglicherweise fehlerhaft ist, oder die NMPA dies verlangt, sollte der MAH das Medizinprodukt neu bewerten und auf der Grundlage der Neubewertungsergebnisse geeignete Risikokontrollmaßnahmen ergreifen. Wenn die Ergebnisse der Neubewertung zeigen, dass die Medizinprodukte Mängel aufweisen, die die persönliche Sicherheit gefährden, und die Risiken nicht durch technische Verbesserungen, Änderungen der Gebrauchsanweisung und der Kennzeichnung beseitigt oder kontrolliert werden können oder das Nutzen-Risiko-Verhältnis inakzeptabel ist, sollte der Zulassungsinhaber die Initiative ergreifen und die Aufhebung der Registrierungsbescheinigung für das Medizinprodukt beantragen.
Rückruf von Medizinprodukten
Rückrufe von Medizinprodukten werden je nach Schweregrad der Mängel in drei Kategorien eingeteilt

Darüber hinaus gibt es Rückrufe, die von der NMPA angeordnet werden. Wenn die NMPA nach einer Untersuchung und Bewertung feststellt, dass der Medizinproduktehersteller fehlerhafte Medizinprodukte zurückrufen sollte, aber nicht die Initiative dazu ergriffen hat, kann sie den Medizinproduktehersteller anweisen, die Medizinprodukte zurückzurufen.
Regelmäßiger Risikobewertungsbericht (PRER)
Der periodische Risikobewertungsbericht (PRER) ist ein wichtiges Dokument für Medizinprodukte. Der MAH muss den PRER des Vorjahres innerhalb von 60 Tagen nach jedem vollen Jahr für Medizinprodukte ausfüllen, die für die Registrierung oder die erstmalige Einreichung in China zugelassen sind. Insbesondere die Zulassungsinhaber von Medizinprodukten der Klassen II und III müssen die PRER über das National Medical Device Adverse Event Monitoring Information System einreichen, das Online-Formular ausfüllen und den PRER als Anhang hochladen. PRER für Medizinprodukte der Klasse I müssen für eine spätere Überprüfung in den Akten aufbewahrt werden. Vor der Erneuerung der Registrierung von Medizinprodukten der Klassen II und III muss der Zulassungsinhaber die PRER für den aktuellen Registrierungszyklus ausfüllen.
Während des gesamten Lebenszyklus des Medizinprodukts sollten die Daten kontinuierlich und ohne Unterbrechung vorgelegt werden. Zu den Datenquellen gehören z. B. Beschwerden, Daten aus Vigilanzdatenbanken und Literatur usw.
Der Hauptinhalt der PRER umfasst:
- Grundlegende Produktinformationen: Name des Medizinprodukts, Modell, Spezifikationen, Nummer der Zulassungsbescheinigung, Aufbau und Zusammensetzung, Hauptbestandteile, Verwendungszweck, Gültigkeitsdauer usw.
- Status der in- und ausländischen Registrierung,
- Maßnahmen zur Risikokontrolle, z. B. Einstellung der Produktion und des Verkaufs des Produkts, Produktrückruf, Prozessverbesserung, Design, technische Anforderungen an das Produkt und Durchführung einer Neubewertung des Medizinprodukts.
- Informationen über unerwünschte Ereignisse, einzelne unerwünschte Ereignisse und Gruppen von unerwünschten Ereignissen, die während des Berichtszeitraums beim Zulassungsinhaber eingegangen sind.
- Zusätzliche Risikoinformationen, z. B. Daten aus Literaturrecherchen, Produktrisikobewertung, Schlüsselüberwachung und Reevaluierung.
- Produktrisiko-Analyse. In diesem Abschnitt sollte die umfassende Produktrisikosituation unter den Gesichtspunkten der Produktauslegung und -entwicklung, des Produktionsmanagements, des Transports und der Lagerung, des Betriebs und der Anwendung, der Wartung, des Kundendienstes usw. analysiert werden, wobei der Schwerpunkt auf der Analyse der Hauptursachen unerwünschter Ereignisse, der Frage, ob sich die Merkmale unerwünschter Ereignisse verändert haben, ob die Häufigkeit der Meldung unerwünschter Ereignisse zugenommen hat, und der Auswirkungen der veränderten Merkmale liegt.
- Schlussfolgerung. Die Unterschiede zwischen den PRER-Ergebnissen und früheren Berichten müssen aufgezeigt werden, ebenso wie die Akzeptanz der Unterschiede. Außerdem sollten die Risikomanagementmaßnahmen zusammengefasst und ihre Notwendigkeit erläutert werden.
qtec als Ihr Partner
Profitieren Sie von der Expertise unserer chinesischen Experten (Muttersprachler), die immer auf dem neuesten Stand der chinesischen Anforderungen sind und Ihnen bei der Erstellung von PMS für China helfen können.

Unser Newsletter „qonzentrat“
kompakt, professionell und präzise
Profitiere von unserem Fachwissen.
 Jetzt anmelden
Jetzt anmelden
 in der Medizintechnik nach GAMP 5 4")

 8")
{kind=link}
{kind=link}
{kind=link}