
Leistungsbewertung unter der IVDR
25. November 2022Unterschiede der IVDR zu den Anforderungen der IVDD
Der Countdown läuft nun auch für alle In-vitro-Diagnostika (IVDs) für den Übergang von der IVDD (Richtlinie 98/79/EG) zur IVDR (Verordnung (EU) 2017/746). Für die meisten Hersteller gibt es hier noch viel zu tun, um alle Anforderungen erfüllen zu können. Gleichzeitig gibt es noch diverse Unklarheiten und Unstimmigkeiten im Verständnis der einzelnen Bestimmungen sowie deren Auslegung für verschiedene Produkte.
Eine entscheidende Frage stellt sich jedem Hersteller bereits bestehender Produkte: Wo genau liegen denn nun die Unterschiede zu den Anforderungen der IVDD? Auf der Hand liegt, dass es sich nun um eine Verordnung und keine Richtlinie mehr handelt. Weiterhin ist die IVDR um einiges umfangreicher geworden – statt 24 Artikel gibt es nun 113. Einige Punkte wurden verschärft und klargestellt, wie zum Bespiel die explizite Aussage, dass die Technische Dokumentation einen Leistungsbewertungs-Plan und -Bericht enthalten muss. Risikobasierte Aspekte sind mehr in den Vordergrund gerückt. Streng genommen wurden allerdings in Bezug auf die Leistungsbewertung bereits unter der IVDD diverse GHTF- (z.B. GHTF/SG5/N6:2012 “Clinical Evidence for IVD medical devices – Key Definitions and Concepts”; GHTF/SG5/N7:2012 “Clinical Evidence for IVD medical devices – Scientific Validity Determination and Performance Evaluation”; GHTF/SG5/N8:2012 “Clinical Evidence for IVD Medical Devices – Clinical Performance Studies for In Vitro Diagnostic Medical Devices”) und CLSI-Dokumente sowie die EN 13612:2002 als Stand der Technik angesehen. Hat man sich als Hersteller bereits daran orientiert, ist das Arbeitspaket für den Übergang zur IVDR deutlich überschaubarer. Allerdings kommt für viele Hersteller durch die Einführung eines neuen risikobasierten Klassifizierungssystems hinzu, dass die erforderliche Dokumentation nun durch eine Benannte Stelle bewertet werden muss.
1. Durchführung einer Leistungsbewertung
Der Leistungsbewertung als wesentlichem Bestandteil des Lebenszyklus eines IVDs kommt eine zentrale Rolle zu. Auch wenn IVDs im Gegensatz zu den klassischen Medizinprodukten generell nicht in direktem Kontakt mit den Patient*innen stehen, kann eine ungenügende Leistung dennoch verheerende Folgen haben, beispielsweise im Falle falsch positiver oder falsch negativer Testergebnisse.
Die Verordnung (EU) 2017/746 (IVDR) verpflichtet den Hersteller, eine Leistungsbewertung für jedes IVD zu erstellen, welches auf dem europäischen Markt in den Verkehr gebracht werden soll. Die Leistungsbewertung wird entsprechend dem Artikel 56 und Anhang XIII, Teil A geplant, durchgeführt und dokumentiert. Das Ziel einer solchen Bewertung besteht darin, die Sicherheit und Leistung des IVDs gemäß der grundlegenden Sicherheits- und Leistungsanforderungen des Anhang I der IVDR („General Safety and Performance Requirements“; GSPR 1-9) entsprechend dem vom Hersteller vorgesehenen Verwendungszweck nachzuweisen. Der Zweckbestimmung (Anhang II, Abschnitt 1.1.c) kommt daher eine entscheidende Rolle zu und es ist deshalb von großer Bedeutung, diese bereits zu Beginn des Entwicklungsprozesses möglichst genau festzulegen.
Der Nachweis, dass das IVD den anwendbaren GSPRs entspricht, erfolgt auf der Grundlage von Daten zur wissenschaftlichen Validität, zur Analyseleistung und zur klinischen Leistung, die demzufolge belegen müssen, dass das Produkt sicher ist und den beabsichtigten klinischen Nutzen erfüllt. Hierbei sollen der Umfang und die Gründlichkeit der Bewertung „verhältnismäßig und angemessen“ sein in Bezug auf die Merkmale des Produktes – einschließlich der Risiken, Risikoklasse, Leistung und Zweckbestimmung. Dabei stellt sich bereits für viele Produkte die schwierige Frage, welcher Umfang denn nun im Einzelfall „angemessen“ und welche Art von Daten erforderlich ist. Ein Guidance Document, auf das sich der Hersteller bei der Erfüllung der Anforderungen an den klinischen Nachweis gemäß IVDR stützen kann, ist die MDCG 2022-2 – „Guidance on general principles of clinical evidence for In Vitro Diagnostic medical devices“.
1.1 Wissenschaftliche Validität
„Wissenschaftliche Validität eines Analyten bezeichnet den Zusammenhang eines Analyten mit einem bestimmten klinischen oder physiologischen Zustand.“ *Quelle: IVDR+
Es soll also wissenschaftlich nachgewiesen werden, dass z.B. ein bestimmter Analyt in ausreichendem Maße geeignet ist, zur Identifizierung einer bestimmten Krankheit herangezogen werden zu können. Dies wird nun auch für Produkte verlangt, die bereits unter der IVDD in Verkehr gebracht wurden. Zu diesem Zweck können wissenschaftliche Literatur, Expertengutachten und technische Standards herangezogen werden. Zusätzlich kann es erforderlich sein, Studien zum Nachweis des Wirkprinzips und / oder klinische Leistungsstudien durchzuführen. Es werden ein Plan sowie ein Bericht erstellt, in denen die wissenschaftliche Validität überprüft und dokumentiert wird (SVP / SVR = „Scientific Validity“ Plan / Report).
1.2 Analyseleistung
„Analyseleistung bezeichnet die Fähigkeit eines Produkts, einen bestimmten Analyten korrekt nachzuweisen oder zu messen.“ *Quelle: IVDR+
Die Analyseleistung wird z.B. durch Parameter wie die analytische Spezifität, analytische Sensitivität und Reproduzierbarkeit sowie Wiederholbarkeit definiert – wie im Anhang I, Abschnitt 9.1 der IVDR beschrieben. Die Überprüfung dieser Parameter erfolgt im Allgemeinen in analytischen Leistungsstudien. Ebenfalls herangezogen werden können Daten aus Verifikations- und Validierungstests und wissenschaftliche Literatur. Bei neuartigen Markern können auch klinische Leistungsstudien erforderlich sein. Die Dokumentation umfasst jeweils einen Plan und Bericht zur Analyseleistung (APP / APR „Analytical Performance“ Plan / Report).
1.3 Klinische Leistung
„Klinische Leistung bezeichnet die Fähigkeit eines Produkts, Ergebnisse zu liefern, die mit einem bestimmten klinischen Zustand oder physiologischen oder pathologischen Vorgang oder Zustand bei einer bestimmten Zielbevölkerung und bestimmten vorgesehenen Anwendern korrelieren“. *Quelle: IVDR]
Die klinische Leistung beschreibt also die Anwendbarkeit des IVDs im klinischen Kontext. Hierbei spielen Parameter wie z.B. diagnostische Spezifität, diagnostische Sensitivität, positiver / negativer prädiktiver Wert und „Likelihood-Verhältnis“ (Anhang I, Abschnitt 9.1) eine wichtige Rolle. Zum Nachweis herangezogen werden können: Daten aus klinischen Leistungsstudien, diagnostischen Routineuntersuchungen und wissenschaftlicher Literatur. Diese werden im klinischen Leistungsbericht dargestellt und bewertet (CPP / CPR = „Clinical Performance“ Plan / Report). Auf klinische Leistungsstudien kann verzichtet werden, wenn ausreichend klinische Daten aus anderen Quellen, z.B. der wissenschaftlichen Literatur oder PMS-Daten, vorliegen. Dies ist im Detail zu begründen.
1.4 Literaturrecherche
Wie bereits erwähnt, können vor allem für den Nachweis der wissenschaftlichen Validität sowie der klinischen Leistung Daten aus der wissenschaftlichen Literatur herangezogen werden. Hierzu wird eine systematische Literatursuche durchgeführt und dokumentiert, vorzugsweise in einem separaten Literatursuche-Plan oder -Protokoll und -Bericht (LSP / LSR), entsprechend der Methodik wie in der MEDDEV 2.7/1, Rev. 4 beschrieben. Die ermittelten Daten werden jeweils im Kontext des medizinischen Hintergrundes und des Stands der Technik bewertet.
Eine Literaturrecherche kann auch bereits im Anfangsstadium der Produktentwicklung sinnvoll sein, um mögliche Risiken anhand vergleichbarer Produkte sowie den Stand der Technik frühzeitig festzustellen.
1.5 Dokumentation
Der erste Schritt für die Leistungsbewertung eines IVDs ist idealerweise die Erstellung eines Leistungsbewertungs-Plans („Performance Evaluation Plan“; PEP) durch den Hersteller. Dieser Plan sollte die in der IVDR, Anhang XIII, Teil A, Abschnitt 1.1 aufgeführten Informationen enthalten – wie die Merkmale und Leistungen des Produktes sowie die Verfahren und Kriterien, die für die Erbringung des erforderlichen klinischen Nachweises angewandt werden.
Der PEP bildet die Grundlage für den PER („Performance Evaluation Report“; PER). Dieser enthält die Ergebnisse der im PEP festgelegten Methoden sowie die Daten zur wissenschaftlichen Validität, der Analyseleistung und der klinischen Leistung gemäß Anhang XIII, Teil A, Abschnitt 1.3.2 und Anhang II, Abschnitt 6.2, einschließlich einer Bewertung dieser Daten. Weitere Aspekte, auf die der PER eingehen sollte, sind z.B. eine Argumentation für die angewandten Methoden zur Erhebung klinischer Nachweise, eine Beschreibung der Zweckbestimmung und damit verbundenen klinischen Claims, eine Bewertung der klinischen Nachweise im Kontext des aktuellen Stands der Technik sowie von Daten aus der Nachbeobachtung bzw. Überwachung nach dem Inverkehrbringen. Ebenso wichtig ist es, dass das Risikomanagement und die Leistungsbewertung eng miteinander verknüpft und aufeinander abgestimmt sind. Die Annehmbarkeit des Nutzen-Risiko-Verhältnisses stellt ebenfalls einen entscheidenden Teil der Bewertung dar. PEP und PER sind wichtige Bestandteile der Technischen Dokumentation eines jeden IVDs.
Für die Evaluierung der wissenschaftlichen Validität, Analyseleistung und klinischen Leistung werden jeweils ein Plan und ein Bericht erstellt (SVP / SVR = „Scientific Validity“ Plan / Report; APP / APR „Analytical Performance“ Plan / Report; CPP / CPR = „Clinical Performance“ Plan / Report). Es scheint von den Benannten Stellen aber auch akzeptiert zu werden, dass diese Pläne und Berichte in den PEP bzw. PER des IVDs integriert werden. Praktikabler ist es allerdings in den meisten Fällen, jeweils separate Pläne und Berichte zu erstellen und diese dann mit der Evaluierung der Daten im PER zu verknüpfen. Kann mit bestimmten Produkten keine Analyseleistung oder klinische Leistung erzielt werden oder sind bestimmte Leistungsanforderungen nicht anwendbar, muss dies im PEP sowie den entsprechenden Einzelberichten ausreichend begründet werden.
Die nachfolgende Abbildung stellt beispielhaft dar, welche Dokumente im Zuge der Erstellung der Leistungsbewertung eines IVDs eine wichtige Rolle spielen. Weiterhin fließen zusätzliche Berichte der Technischen Dokumentation, wie zum Beispiel die Risikomanagement-Akte sowie die Bewertung der Biokompatibilität, in die Leistungsbewertung ein.

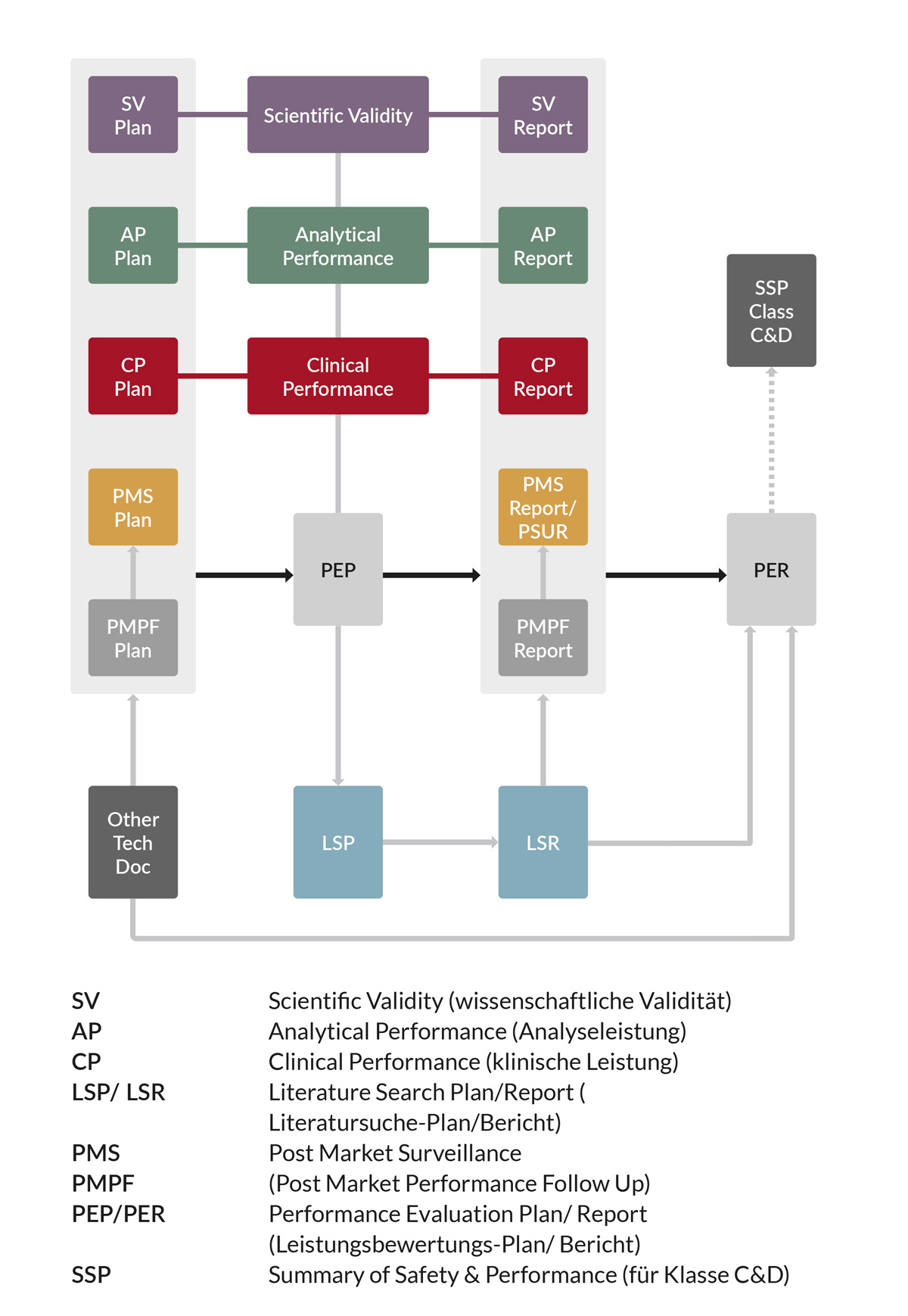

2. Kurzbericht über Sicherheit und Leistung für Klasse C und D
Für Produkte der Klassen C und D müssen die Hersteller die wichtigsten Sicherheits- und Leistungsaspekte des Produktes sowie das Ergebnis der Leistungsbewertung in einem Dokument zusammenfassen („Summary of safety and performance“; SSP) und über EUDAMED öffentlich zugänglich machen. Der Bericht umfasst die in Artikel 29 aufgeführten Inhalte und ist regelmäßig zu aktualisieren. MDCG 2022-9 „Summary of safety and performance template“ schlägt eine Vorlage für den SSP vor.
Unsere Fortbildungen
Sie möchten mehr erfahren? Dann buchen Sie das passende Seminar zum Thema. Oder benötigen sie eine Inhouse-Schulung? Dann kontaktieren Sie uns gerne unter academy@qtec-group.com

3. Lebenszyklus
Leistungsbewertung ist ein kontinuierlicher Prozess. Der klinische Nachweis und seine Bewertung im PER werden während des gesamten Lebenszyklus des IVDs aktualisiert. Wichtige Daten liefern hierbei die Nachbeobachtung der Leistung gemäß PMPF-Plan („Post Market Performance Follow Up“; Anhang XIII, Teil B) sowie die Überwachung nach dem Inverkehrbringen gemäß PMS-Plan („Post Market Surveillance“; Artikel 79). „Wird die Nachbeobachtung der Leistung nach dem Inverkehrbringen für ein bestimmtes Produkt nicht für angemessen gehalten, so wird im Bericht über die Leistungsbewertung eine Begründung angegeben und dokumentiert.“ *Quelle: IVDR
Wie häufig der PER aktualisiert werden muss, hängt von der Risikoklasse des Produktes ab. Produkte der Klassen A und B können nach Bedarf aktualisiert werden, ein Zyklus von mindestens drei Jahren wird allerdings empfohlen. IVDs der Klassen C und D werden ebenfalls je nach Bedarf aktualisiert, jedoch mindestens einmal jährlich.
4. Fazit und Stolpersteine
Die Leistungsbewertung soll belegen, dass ein IVD sicher ist und den vom Hersteller beabsichtigten klinischen Nutzen erfüllt. Dieser Nachweis erfolgt auf der Grundlage von Daten zur wissenschaftlichen Validität, zur Analyseleistung und zur klinischen Leistung. Umfang und Art der erforderlichen Daten variieren dabei stark zwischen verschiedenen Produkten, in Abhängigkeit von Risiken, Risikoklasse, Leistung und Zweckbestimmung. Wichtig ist in jedem Fall, in ausreichendem Maße zu argumentieren, warum im Einzelfall auf bestimmte Studien, Nachweise bzw. Daten verzichtet wird. Leistungsbewertungsplan (PEP) sowie Leistungsbewertungsbericht (PER) stellen einen essenziellen Teil der Technischen Dokumentation dar.
Nach wie vor gibt es diverse Hürden bei der Umsetzung der IVDR zu überwinden. Fragestellungen und Herausforderungen sind für jedes Produkt anders. Welcher Umfang und welche Art von Daten sind erforderlich, um die wissenschaftliche Validität, Analyseleistung und klinische Leistung eines IVDs nachzuweisen? Welche Quellen kann man hierzu heranziehen? Ist eine Leistungsstudie erforderlich? Und wenn ja, ist das geplante Studiendesign angemessen? …
Herausforderungen bestehen aber teilweise auch in Gegebenheiten, die nicht vom Hersteller beeinflusst werden können. So gibt es zum Beispiel Schwierigkeiten, EU-Referenzlaboratorien (EURL) für die vorgeschriebene Prüfung für IVDs der Klasse D (Artikel 48, Absatz 5) in ausreichender Anzahl sowie auch Expertinnen und Experten für die Konsultation (Artikel 48, Absatz 6) zu gewinnen. Ebenso kann es schwierig sein, eine Benannte Stelle für die Konformitätsbewertung zu finden – sofern bisher keine involviert war.
Welcher Art Ihre Herausforderungen bei der Umsetzung der IVDR auch sein mögen, wir lassen Sie nicht allein! Bei der Planung, Durchführung und Dokumentation Ihrer Leistungsbewertung unterstützen wir Sie gerne.
Lesen Sie zum Thema IVDR auch unsere folgenden Blogartikel und Detailseiten:
Wir können nicht nur Clinical Affairs
Fragen Sie unsere Expertinnen und Experten nach den besten Tipps zur Erstellung einer Leistungsbewertung. Welcher Umfang angemessen ist und ob der Nachweis zur wissenschaftlichen Validität ausreichend ist, können wir in einem gemeinsamen Meeting erörtern.

Unser Newsletter „qonzentrat“
kompakt, professionell und präzise
Profitiere von unserem Fachwissen.
 Jetzt anmelden
Jetzt anmelden
 in der Medizintechnik nach GAMP 5 4")

 8")
{kind=link}
{kind=link}
{kind=link}