
IVDs unter der IVDR: Klassifizierung und Konformitätsbewertung
17. November 2022
Höhere und komplexere Anforderungen unter der IVDR (in vitro diagnostic)
Im Mai 2022 endete die Übergangsfrist der bisherigen Richtlinie 98/79/EG über In-vitro-Diagnostika (IVDD / in vitro diagnostic). Damit schloss sich das letzte Hintertürchen für ggf. eine erleichterte Marktzulassung für IVDs. Seitdem greift der volle Umfang der EU-Verordnung 2017/746 über In-vitro-Diagnostika (IVDR). Sehr viele Hersteller von IVD-Produkten sehen sich unter der IVDR plötzlich deutlich höheren und komplexeren Anforderungen ausgesetzt als vorher. Das hat vor allen Dingen zwei Gründe:
- Anstelle der einfachen Eingruppierung anhand von Listen, wie sie in der IVDD stattgefunden hat, muss nun eine komplexe Klassifizierung anhand von 7 Regeln erfolgen.
- Für viele Produkte, für die vorher keine Einbindung einer Benannten Stelle (BS) erfolgen musste, wird sie nun obligatorisch. Die Hersteller müssen sich nun mit den verschiedenen möglichen Konformitätsbewertungsverfahren und Sonderregelungen für bestimmte Produktgruppen auseinandersetzen.
Zweckbestimmung von IVDs
Die Klassifizierung richtet sich nach der Zweckbestimmung des Produktes. Generell gilt es folgendes zu beachten:
Schritt 1:
Zunächst muss ein Produkt als IVD qualifiziert werden. Dazu muss die Definition eines IVDs (Artikel 2, Absatz 2) erfüllt sein:
„In-vitro-Diagnostikum“ bezeichnet ein Medizinprodukt, das als Reagenz, Reagenzprodukt, Kalibrator, Kontrollmaterial, Kit, Instrument, Apparat, Gerät, Software oder System — einzeln oder in Verbindung miteinander — vom Hersteller zur In-vitro-Untersuchung von aus dem menschlichen Körper stammenden Proben, einschließlich Blut- und Gewebespenden, bestimmt ist und ausschließlich oder hauptsächlich dazu dient, Informationen zu einem oder mehreren der folgenden Punkte zu liefern,
-
- über physiologische oder pathologische Prozesse oder Zustände,
- über kongenitale körperliche oder geistige Beeinträchtigungen,
- über die Prädisposition für einen bestimmten gesundheitlichen Zustand oder eine bestimmte Krankheit,
- zur Feststellung der Unbedenklichkeit und Verträglichkeit bei den potenziellen Empfängern,
- über die voraussichtliche Wirkung einer Behandlung oder die voraussichtlichen Reaktionen darauf oder
- zur Festlegung oder Überwachung therapeutischer Maßnahmen.
Probenbehältnisse gelten als auch In-vitro-Diagnostika.
Schritt 2:
Erfüllt das Produkt diese Definition nicht, gilt es zu prüfen, ob es sich um ein Zubehör eines IVDs handelt. Dieses ist wie folgt definiert:
„Zubehör eines In-vitro-Diagnostikums“ bezeichnet einen Gegenstand, der zwar an sich kein In-vitro-Diagnostikum ist, aber vom Hersteller dazu bestimmt ist, zusammen mit einem oder mehreren bestimmten In-vitro-Diagnostika verwendet zu werden, und der speziell dessen / deren Verwendung gemäß seiner / ihrer Zweckbestimmung(en) ermöglicht oder mit dem die medizinische Funktion des In-vitro-Diagnostikums / der In-vitro-Diagnostika im Hinblick auf dessen / deren Zweckbestimmung(en) gezielt und unmittelbar unterstützt werden soll;
Schritt 3:
Ein Blick in Artikel 1 gibt Aufschluss über den Geltungsbereich der IVDR. Hier werden ebenfalls Ausschlusskriterien für die Anwendbarkeit der IVDR festgelegt. So heißt es:
Diese Verordnung gilt nicht für:
- Produkte für den allgemeinen Laborbedarf oder allein für Forschungszwecke bestimmte Produkte, es sei denn, sie sind aufgrund ihrer Merkmale vom Hersteller speziell für In-vitro-Untersuchungen bestimmt;
- invasive zur Entnahme von Proben bestimmte Produkte oder Produkte, die zum Zweck der Probenahme direkt am menschlichen Körper angewendet werden;
- auf internationaler Ebene zertifizierte Referenzmaterialien;
- Materialien, die für externe Qualitätsbewertungsprogramme verwendet werden.
Fällt das Produkt unter die Definition eines IVDs oder eines Zubehörs eines IVDs und ist nicht aus dem Geltungsbereich der IVDR ausgeschlossen, so wird das Produkt nun den strengen Kriterien des Anhang VIII, IVDR unterworfen – den Klassifizierungsregeln.

Klassifizierung nach Anhang VIII
Das neue regelbasierte Risikoklassifizierungssystem der IVDR ist flexibler als das listenbasierte System der IVDD, das es ersetzt. Die IVDR ermöglicht es, besser mit dem technologischen Fortschritt und der Notwendigkeit, sich mit neuen Erkrankungen zu befassen, Schritt zu halten. Anstatt bestimmte IVD-Produkte oder Erkrankungen zu benennen, richtet sich die Risikoklassifizierung eines Produkts nach seiner Zweckbestimmung und berücksichtigt nicht nur das Risiko für den Einzelnen, sondern auch das Risiko für die öffentliche Gesundheit. Um sein IVD zu klassifizieren, sollte der Hersteller die in Anhang VIII der Verordnung aufgeführten Regeln anwenden.
Anhang VIII der IVDR teilt sich in 2 Teile auf:
Teil 1 mit allgemeinen Vorschriften zur Klassifizierung, wie z.B.:
- Die Anwendung der Klassifizierungsregeln richtet sich nach der Zweckbestimmung der Produkte. Die Festlegung der Zweckbestimmung für ein IVD gibt Ihnen als Hersteller regulatorische Freiräume. Ein Beispiel: Die Zweckbestimmung kann so definiert werden, dass ein Produkt aus mehreren Komponenten besteht. In diesem Fall kann das Gesamt-Produkt als ein IVD klassifiziert und zugelassen werden (Option A). Alternativ kann jede Komponente für sich eine einzelne Zweckbestimmung erhalten, einzeln klassifiziert und zugelassen werden (Option B). Einzelzulassungen sind dann von Vorteil, wenn zum Beispiel die Einzelkomponenten unterschiedlich klassifiziert werden würden, denn die Durchführungsvorschrift in Anhang VIII, Absatz 1.9 besagt, wenn mehr als eine Regel zutrifft, sollte die Regel befolgt werden, die zur höchsten Einstufung führt. Somit würden nicht alle Komponenten in eine höhere Risikoklasse fallen, sondern nur Teile.

Teil 2: Die 7 Klassifizierungsregeln
Jede der 7 Klassifizierungsregeln ist für ein IVD Produkt auf Anwendbarkeit zu prüfen. Die zutreffende Regel definiert die Risikoklasse. In Übereinstimmung mit den internationalen Klassifizierungsprinzipien sind diese vier Klassen definiert:
- geringes individuelles Risiko und geringes Risiko für die öffentliche Gesundheit
- moderates individuelles Risiko und / oder geringes Risiko für die öffentliche Gesundheit
- hohes individuelles Risiko und / oder moderates Risiko für die öffentliche Gesundheit
- hohes individuelles Risiko und hohes Risiko für die öffentliche Gesundheit.
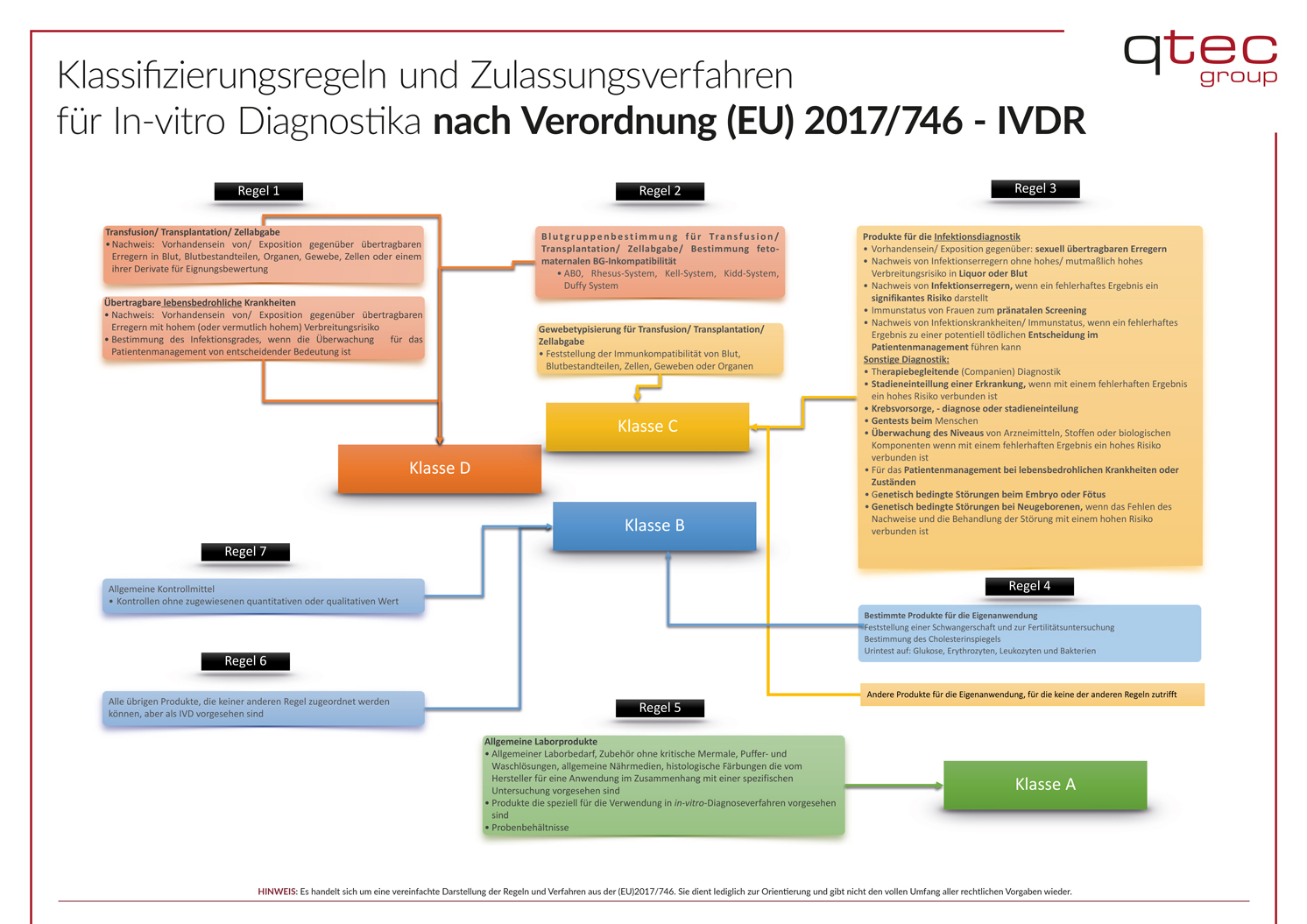
Die Klassifizierung eines IVDs obliegt in erster Linie dem Hersteller (Artikel 2, Abs. 23). Unstimmigkeiten zwischen der BS und dem Hersteller bezüglich der Klassifizierung werden von der zuständigen Behörde geklärt. Die zuständige Behörde ist die Behörde des Mitgliedstaats, in dem der Hersteller oder der autorisierte Bevollmächtigte (Artikel 2, Abs. 25) seine eingetragene Niederlassung hat. Zwei Behörden können beteiligt werden, wenn der Hersteller und die BS ihren Sitz in unterschiedlichen EU-Ländern haben und daher unterschiedlichen Behörden unterstehen (Artikel 47). De EU Kommission gibt Ihnen weitere Hilfestellungen bei Klassifizierungsfragen:
- MDCG 2020-16 - Guidance on Classification Rules for in vitro Diagnostic Medical Devices under Regulation (EU) 2017/746.
- Manual on borderline and classification for medical devices under Regulation (EU) 2017/745 on medical devices and Regulation (EU) 2017/746 on in vitro diagnostic medical device.
- MDCG 2019-11 - Qualification and classification of software - Regulation (EU) 2017/745 and Regulation (EU) 2017/746.
Konformitätsbewertungsverfahren
Das Konformitätsbewertungsverfahren eines IVD-Produkts variiert je nach Risikoklasse und spezifischen Merkmalen bestimmter Produkte (Artikel 48). Die Benannte Stelle ist für alle Produkte der Klassen B, C und D sowie für sterile Produkte der Klasse A erforderlich (Artikel 48, Absatz 10). Die verschiedenen Bewertungswege je nach Produktklasse sind in Artikel 48 sowie den Anhängen IX, X, XI beschrieben. In einigen Fällen haben die Hersteller eine Wahl hinsichtlich des Konformitätsbewertungsweges. Eine Übersicht finden Sie hier:
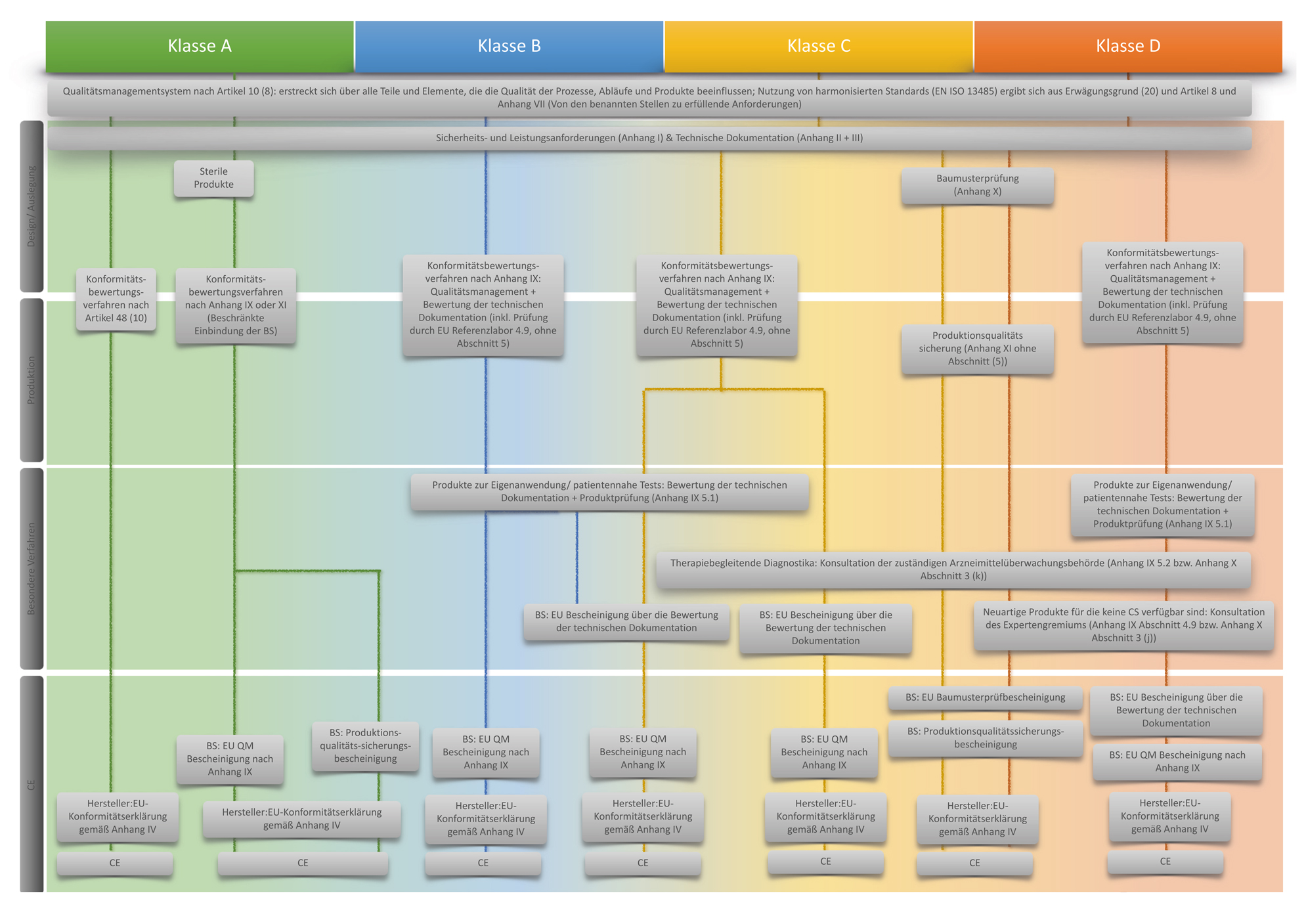
Neben der Bewertung durch eine BS, was kommt on top?
Therapiebegleitende IVDs (sogenannte Companion Diagnostics) durchlaufen ein Konsultationsverfahren bei der Arzneimittelüberwachungsbehörde (Artikel 48, Absatz 4).
Besondere Herausforderungen erwarten Hersteller von IVD-Produkten der Klasse D. Die Konformitätsbewertung von Produkten der Klasse D erfordert zusätzlich die Einbeziehung eines EU-Referenzlabors, um die vom Hersteller definierten Leistungen und die Übereinstimmung mit den anwendbaren Gemeinsamen Spezifikationen (CS) (Artikel 48, Absatz 5) zu überprüfen. Die aktuellen CS für bestimmte Klasse D IVDs sind in der Durchführungsverordnung (EU) 2022/1107 beschrieben.
EU-Referenzlaboratorien sind wissenschaftliche Laboratorien innerhalb der Europäischen Union, die darauf ausgelegt sind, die Sicherheit und Konformität von IVD mit der Verordnung zu gewährleisten. Diese EU-Referenzlaboratorien sind in der der Durchführungsverordnung (EU) 2023/2713 benannt.
Neuartige IVDs der Klasse D, für die keine CS definiert sind, durchlaufen ein weiteres Konsultationsverfahren durch ein unabhängiges Expertengremium der EU-Kommission (Artikel 48, Absatz 6). MDCG 2021-22 erklärt genauer, was als neuartig angesehen wird. Auch wenn die Koordination von EU-Referenzlaboratorien und Konsultationsverfahren nicht die Aufgabe der Hersteller ist, sollten Sie die Stationen des Konformitätsbewertungsverfahrens kennen, da sie zeitaufwendig und ggf. kostspielig sind.
Fazit
Mit der Implementierung der IVDR wurden die Anforderungen an IVDs gewaltig nach oben geschraubt. Die signifikante Änderung bei der Klassifizierung von IVDs führt bei vielen Produkten zu einer höheren Risikoklasse, welche die Einbeziehung einer BS bedingt. Die EU-Kommission schätzt, dass rund 85 % aller IVDs durch die Neuklassifizierung von den BS zertifiziert werden müssen. Ein Großteil der Produkte erfährt dieses Prozedere zum ersten Mal. Eine strategisch kluge Zusammenfassung von Komponenten bzw. Abgrenzung von Einzelprodukten und damit einhergehend die Festlegung der Zweckbestimmung kann Ihnen bei Klassifizierung und Produktzulassung Vorteile bringen. Sprechen Sie uns gerne an. Wir unterstützen Sie auf Ihrem Weg der Zulassung.
Zulassung von IVDs unter der IVDR
Die Klassifizierung und Konformitätsbewertung von IVDs unter der IVDR ist eine Wissenschaft für sich. Unsere Expertinnen und Experten haben den Überblick und unterstützen Sie bei der Zulassung. Sprechen Sie uns an.

Unser Newsletter „qonzentrat“
kompakt, professionell und präzise
Profitiere von unserem Fachwissen.
 Jetzt anmelden
Jetzt anmelden
 in der Medizintechnik nach GAMP 5 4")

 8")
{kind=link}
{kind=link}
{kind=link}